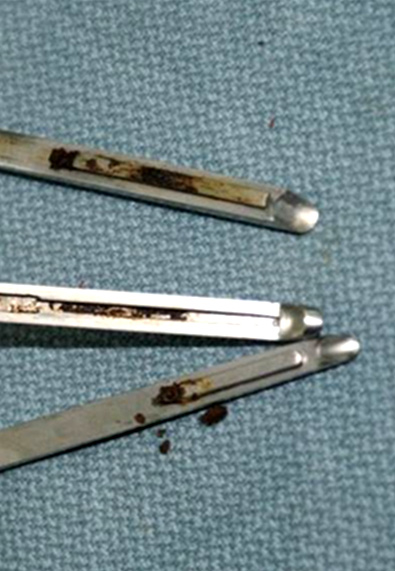
These are disassembled Kerrison Rongeurs that were reprocessed using the manufacturer’s non-validated IFUs.
(And Why They Are So Important)
The FDA mandates that all reusable medical device manufacturers provide instructions for reprocessing their devices (see 7/21/17 Novo blog). These instructions are called IFUs and they provide reprocessing personnel with the necessary information and steps to safely reprocess a reusable medical device. Included in every IFU are specific instructions on how to properly decontaminate and clean the device prior to sterilization.
Medical device reprocessing personnel have always been told, “If it’s not clean, it can’t be sterile.” The more accurate statement is “If it’s not clean, it can’t be safe!” Just because you sterilize a device does not mean that it is safe to use, especially if it is still contaminated with residual bio-burden prior to sterilization. A frequent cause of a surgical infection is dead, sterile bio-burden that gets deposited into the body from a ‘sterile’ instrument that was not completely clean prior to sterilization.
Over the past forty years, the FDA has taken an ever increasing interest in reusable device IFUs and their impact on patient safety. The FDA’s concern with the content and the validity of manufacturers’ IFUs was codified in March of 2015 when the FDA issued “Reprocessing Medical Devices in Health Care Settings: Validation Methods and Labeling."
In Section VII of the FDA report, Validation of Reprocessing Methods in Accordance with the Quality System Regulation, the report states:
For class II and class III devices and select class I devices, manufacturers must establish and maintain procedures for validating the design of their device, which shall ensure that the device conforms to defined user needs and intended uses. 21 CFR 820.30(g). Manufacturers must also establish and maintain procedures for monitoring and control of process parameters for validated processes to ensure that the specified requirements continue to be met, 21 CFR 820.75(b). Establishing procedures includes implementation. 21 CFR 820.3(k). FDA interprets these regulations to require manufacturers to validate the design, including reprocessing instructions, of reusable devices to ensure that the device can be effectively reprocessed and safely reused over its use life, as intended.
The ‘dirty’ little secret in our industry is that the FDA did not make the new requirement for manufacturers to validate their IFUs retroactive. Specifically, at present, the FDA’s IFU validation requirement only applies to new product 510(k) submissions to the FDA. Regretfully, almost all of the tens-of-thousands of surgical instruments that are used in patients every day have never had their cleaning and their sterilization IFUs validated.
“It’s the sad reality of what people in CS/SPD face — they are held accountable for returning clean, sterile, moisture-free instruments up to the OR and yet they are dealing with instruments whose cleaning IFUs have never been validated and they are having to rely on visual inspection, which is not an acceptable technique.”¹
If you are relying on manufacturer IFUs that have not been validated “to ensure that the device can be effectively reprocessed and safely reused,” then you cannot be sure that the instruments are clean, sterile and moisture-free on every reprocessing cycle. The question that every CS/SPD personnel need answer is “Do I know that the instruments that I am sending to the O.R. that will be put inside of our patients are really safe?”
Reducing your patients’ risk of a surgical infection caused by a contaminated instrument starts by only using instruments whose IFUs have been validated using FDA validation testing protocols. You must insist on validated IFUs to protect your patients.
1.“Validated? It’s complicated” by Kara Nadeau, HPN, April 2017
August 4, 2017
5 view(s) 